
SCN3A – a sodium channel in epilepsy and brain malformations
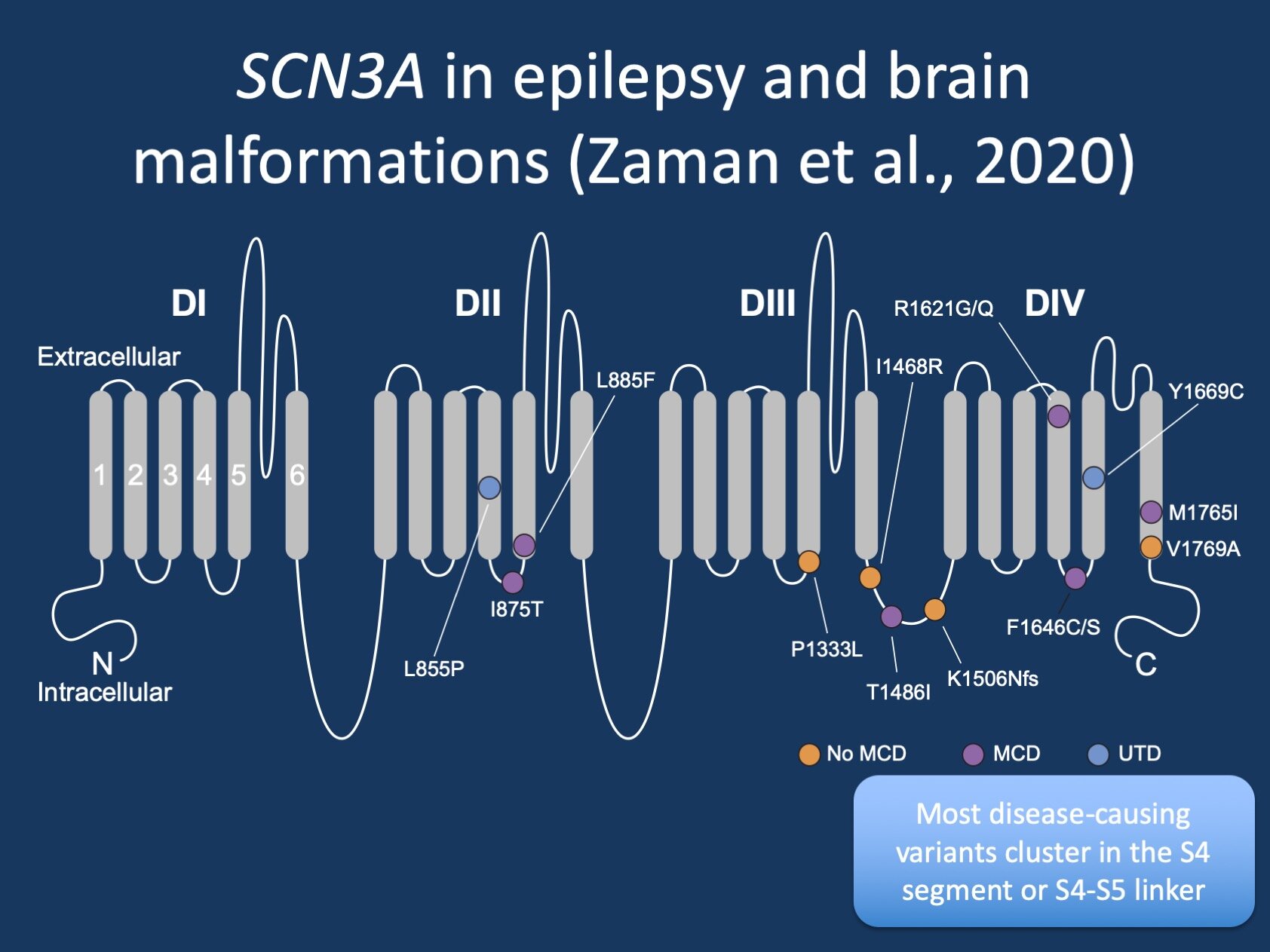
Figure 1. SCN3A in epilepsies and malformations of cortical development. Many variants cluster in the S4 segment or S4/S5 linker region and result in a gain-of-function effect of the Nav1.3 protein, the ion channel encoded by the SCN3A gene (figure modified from a prior version of Zaman et al., 2020).
Posted on June 17th, 2020 by Ingo Helbig
The missing ion channel. A little more than two years ago, we wrote about our discovery of SCN3A as a novel disease gene in epileptic encephalopathies. At the time, SCN3A was the missing ion channel, the only brain-expressed voltage gated sodium channel that did not have a clear gene-disease relationship. However, since the initial discovery of SCN3A as a disease gene, both the phenotypic spectrum and variant landscape have expanded considerably. In a recent publication, we updated our knowledge based on information of 22 individuals with SCN3A-related disorders, which showed brain malformations in more than 75% of individuals and an unusual clustering of pathogenic variants in parts of the Nav1.3 channel.
Nav1.3. I typically start my blog posts on sodium channels with a brief acknowledgement that the nomenclature of sodium channels is confusing, as all channels are named twice. Ion channels have a “physiologic” name (Nav1.3) and a “genetic” name (SCN3A). This divide is historical and one can only speculate than nobody would have expected the fields of ion channel physiology and human genetics to merge to his extent. For the most part, the nomenclature is straightforward with regards to numbering, including Nav1.1 (SCN1A), Nav1.2 (SCN2A), and Nav1.3 (SCN3A). However, the SCN8A gene is an important exception, as it refers to Nav1.6 (SCN8A). As mentioned in our previous post, Nav1.3 encoded by the SCN3A gene is highly expressed in the developing brain, but its expression drops to basically insignificant levels postnatally.
The SCN3A spectrum. In the publication by Zaman and collaborators, we were able to include 22 individuals with SCN3A-related disorders. What becomes clear quite quickly is that SCN3A has some properties that clearly set it apart from the remaining triad of disease-causing neuronal voltage-gated sodium channels (SCN1A, SCN2A, SCN8A). Specifically, there are three aspect that make SCN3A unique.
1 – Brain malformations. While we hinted at this in our initial study, more than 75% of individuals with SCN3A-related disorders have brain malformation (malformations of cortical development). Neuroimaging findings were observed in in 15/19 individuals with available MRIs and showed bilateral diffuse or perisylvian polymicrogyria in most patients. While malformations of cortical development had previously been seen in other channelopathies on rare occasions, SCN3A is the only ion channel that results in brain malformations in the majority of individuals. This fact cannot be over-emphasized: SCN3A completely rearranges our concepts of what channelopathies are. The SCN3A example shows that genetic alterations in ion channels are not only functional changes occurring in an otherwise typically developed Central Nervous System, but can fundamentally alter the way that the cerebral cortex develops.
2 – Gain-of-function. We know from other brain-expressed sodium channelopathies that both gain- and loss-of-function variants can result in disease. SCN2A is the prime example for this, where gain-of-function (GoF) variants result in early-onset epileptic encephalopathies, while loss-of-function (LoF) variants primarily result in neurodevelopmental disorders including autism, often with later-onset epilepsies. Again, SCN3A is different. Nearly all known disease-causing variants in SCN3A are GoF, which means that SCN3A-related disorders are an almost exclusive gain-of-function disease, an observation that is critical for future drug development in this condition. The GoF properties observed with pathogenic SCN3A variants fall into a combination of the following three categories: (1) increased persistent current; (2) hyperpolarizing shift in the voltage dependence of activation; (3) slowed fast inactivation. Essentially, these pathogenic variants affect how the channel opens (too soon), how the channel closes (too slowly), and how much sodium current remains after the initial inward flux of sodium that depolarizes the cell (too much). What remains unclear is why some pathogenic SCN3A variants that show similar GoF properties can cause cortical malformations in some instances but not others.
3 – Variant spectrum. The third unusual feature of SCN3A-related disorders is the spectrum of disease causing variants that cluster in particular parts of the Nav1.3 channel. Most variants are located in the S4 segment or the S4-S5 linker. Knowing that disease-causing variants in SCN3A have specific electrophysiological properties (see above), this allows for some general insights into how sodium channels work. The entire world of sodium channel function has been mapped to a near-atomic level and is unfortunately too much to discuss in this blog post. However, with regards to SCN3A, it is important to point out that these variants occur in regions of the sodium channel that are crucial for changing the structure of the channel while it is opening and closing.
In a nutshell. SCN3A-related disorders challenge our paradigms with regards to channelopathies. Most individuals with SCN3A-related disorders have malformations of cortical development, variants are invariably due to gain-of-functions effects secondary due to the opening/closing properties of the channel. The variant spectrum is unusually restricted with most variants occurring in proximity of the S4 segment. SCN3A-related disorders may be more common than previously suspected and understanding the biological basis may lead to important future steps towards personalized drug development.